日常积累
omics 组学
组学(英语:omics)通常指生物学中对各类研究对象(一般为生物分子)的集合所进行的系统性研究,例如,基因组学、蛋白质组学,和代谢物组学等,而这些研究对象的集合被称为组。在英文中,“组”以**-ome作为后缀,而“组学”以-omics**作为后缀。例如,基因组学(genomics)是系统性研究生物体基因组(genome)中各种基因(gene)以及它们之间的相互关系的学科。
transcriptome转录组
转录组(英语:Transcriptome),也称为“转录物组”,广义上指在相同环境(或生理条件)下的在一个细胞、或一群细胞中所能转录出的所有RNA的总和,包括信使RNA(mRNA)、核糖体RNA(rRNA)、转运RNA(tRNA)及非编码RNA;狭义上则指细胞所能转录出的所有信使RNA(mRNA)。
biological interpretation生物学解释
proteome蛋白质组
蛋白质组(也称蛋白质体,proteome),是在特定时间内,是一个由基因组、细胞、组织、或生物体表达的或可以表达的整套蛋白质。 它是在给定时间,在给定条件下在给定类型的细胞或生物中表达的蛋白质的集合。 研究蛋白质组的学科就是蛋白质组学。
cell membrane细胞膜
细胞膜,又称原生质膜或质膜(英语:cell membrane 或 plasma membrane 或 cytoplasmic membrane),为细胞结构中分隔细胞内、外不同介质和组成成分的界面。原生质膜普遍认为由磷脂质双层分子作为基本单位重复而成,即磷脂双分子层,其上镶嵌有各种类型的膜蛋白以及与膜蛋白结合的糖和糖脂。
next-generation sequencing technology下一代测序NGS
从原理上说,桑格测序与新一代测序(NGS)技术背后的概念是相似的。在NGS和桑格测序(也称为双脱氧测序或毛细管电泳测序)的过程中,DNA聚合酶会将有荧光的核苷酸逐个添加到正在延长的DNA模板链上。通过荧光标记识别每个掺入的核苷酸。
桑格测序与NGS的主要差异在于测序量。桑格测序一次只能对一条DNA片段进行测序,而NGS可以进行大规模平行测序,每次运行可同时对数百万个片段进行测序。这意味着这样的高通量过程一次可对数百到数千个基因进行测序。NGS还具有更强的探索能力,可以通过深度测序检测新型或罕见变异。
human cell atlas project 人类细胞图谱计划
这是一项大型国际合作项目:根据独特的分子信息(如基因表达)对所有人类细胞种类进行定义,并将这些信息与传统的细胞学表述(如位置和形态)相关联。
CPM
counts per million
- cpm: Counts-per-million. This is the read count for each gene in each cell, divided by the library size of each cell in millions.
不同样品的测序量有差异,最简单的标准化方式是计算counts per million (CPM) = 原始reads count ÷ 总reads数 x 1,000,000;
这种计算方式,易受到极高表达且在不同样品中存在差异表达的基因的影响:这些基因的打开或关闭会影响到细胞中总的分子数目,可能导致这些基因标准化之后就不存在表达差异了,而原本没有差异的基因标准化之后却有差异了;
RPKM、FPKM、TPM 是 CPM按照基因或转录本长度归一化后的表达,都会受到这一影响;
- RPM没有考虑转录本的长度的影响。适合于产生的read读数不受基因长度影响的测序方法,比如miRNA-seq测序,miRNA的长度一般在20-24个碱基之间
- RPKM/FPKM考虑了转录本的长度的影响。适用于基因长度波动较大的测序方法,如lncRNA-seq测序,lncRNA的长度在200-100000碱基不等
- TPM是先去除了基因长度的影响,而RPKM/FPKM是先去除测序深度的影响。TPM实际上改进了RPKM/FPKM方法在跨样品间定量的不准确性。
Normalization可以借助许多工具
- Falco [云端处理流程]
- SCONE(Single-Cell Overview of Normalized Expression)—一个质控和标准化的包
SCONE is an R package for comparing and ranking the performance of different normalization schemes for single-cell RNA-seq and other high-throughput analyses.
- Seurat– QC以及后续分析探索数据
- ASAP(Automated Single-cell Analysis Pipeline) —交互式网页分析
CV
CV² = a1/μ + α0
Coefficient of variation
变异的系数
CV = (标准偏差 SD / 平均值Mean )x 100%
The coefficient of variation (CV) is a statistical measure of the dispersion of data points in a data series around the mean. The coefficient of variation represents the ratio of the standard deviation to the mean(标准差/平均值,表示概率分布的离散程度), and it is a useful statistic for comparing the degree of variation from one data series to another, even if the means are drastically different from one another.
The coefficient of variation shows the extent of variability of data in a sample in relation to the mean of the population.
举个小例子:
PC
principle component
RLE
relative log expression
量化因子 (size factor,SF),首先计算每个基因在所有样品中表达的几何平均值;每个细胞的SF是所有基因与其在所有样品中的表达值的几何平均值的比值的中位数;由于几何平均值的使用,只有在所有样品中表达都不为0的基因才能用来计算。这一方法又被称为RLE(relative log expression)。

Poisson distribution 泊松分布
独立且同分布
RNA spike-in
An RNA spike-in is an RNA transcript of known sequence and quantity used to calibrate measurements in RNA hybridization assays, such as DNA microarray experiments, RT-qPCR, and RNA-Seq.
A spike-in is designed to bind to a DNA molecule with a matching sequence, known as a control probe.This process of specific binding is called hybridization. A known quantity of RNA spike-in is mixed with the experiment sample during preparation.The degree of hybridization between the spike-ins and the control probes is used to normalize the hybridization measurements of the sample RNA.
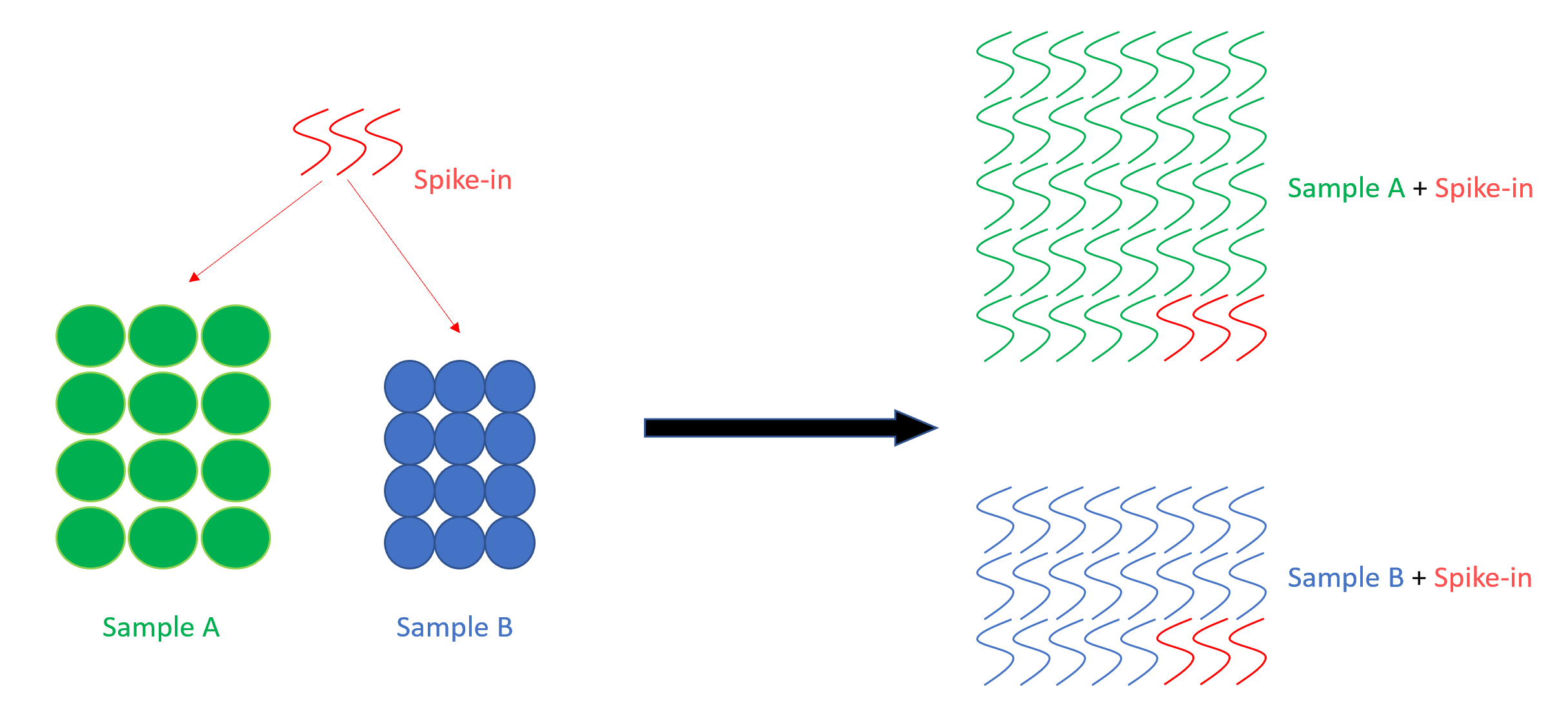
RNA的spike-in是一组序列已知的RNA转录本,目前普遍使用的是ERCC(The External RNA Control Consortium)搞出来的一组RNA序列。当然,也可以使用序列相似度较低的物种的序列作为spike-in。如两种常见的酵母,S.pombe和S.cerevisiae,他们的序列可以作为彼此的spike-in进行后续矫正。
通过在样品制备过程中,混入指定数量的spike-in,我们就可以知道不同样本中的基因绝对比表达值。如等细胞数的样本A和样本B,在每个样本中,我加入了等量的spike-in。最后分析发现,spike-in占样本A的1%,但是占样本B的5%。这表明样本A的RNA表达量也许普遍比样本B的表达量高五倍左右。
下面是一个简单的草图,希望可以帮助理解,左边是细胞,右边是RNA
read counts
(1)数值概念:比对到gene A的reads数。
(2)用途:用于换算CPM、RPKM等后续其他指标;作为基因表达差异分析的输入数值。
大部分差异分析软件(如DESeq和edgeR),用原始的可比对的reads count作为输入,并用负二项分布模型估算样本间基因差异表达的概率。
https://www.jianshu.com/p/0f5a9616efe2
原始测序数据经过处理得到分子计数矩阵(count matrix),或者reads count (读数矩阵)。这取决于单细胞文库构建方案中是否包含唯一分子标识符(UMI, unique molecular identifiers)(参考下面的Box1. Hemberg-lab单细胞转录组数据分析(六)- 构建表达矩阵,UMI介绍)。原始数据处理流程如Cell Ranger, indrops, SEQC, zUMIs负责测序数据质控,确定reads来源的细胞 (barcodes) (这一步也称为demultiplexing)和mRNA分子 (生信宝典注:单细胞测序不只获得mRNA,更准确说是带poly-A尾巴的RNA,包括mRNA和lncRNA等)、基因组比对和定量。获得的reads或count矩阵的行数等于barcodes的数目,列数等于基因数目(生信宝典注:也可能反过来,行为基因,列为barcodes)。这里使用术语barcodes而不是cell,因为分配给相同barcode的所有reads可能并不只是来源于同一细胞。barcode可能会错误地标记多个细胞(doublet),也可能不会标记任何细胞(空液滴/孔) 。
尽管reads data和 count data 的测量噪声级别不同 (生信宝典注:基于UMI的数据,获得的是分子计数,count data,噪声更低),但在分析流程中的处理步骤是相同的。为简单起见,在本教程中,我们将数据称为计数矩阵(count data)。在reads和计数矩阵的结果不同的地方,将会特别提到reads矩阵。
构建文库
该构建方法包括以下步骤:对单细胞中的RNA进行反转录得到cDNA;用扩增引物对cDNA进行预扩增,得到扩增cDNA;对扩增cDNA进行片段化文库构建,得到单细胞的转录组测序文库;其中,扩增引物中的dTTP替换为dUTP。
基因文库library
在分子生物学中, 一个基因文库是通过分子克隆的方法存储和繁殖微生物群的DNA片段的集合。 基因文库有很多种,包括部分基因文库(cDNA文库)、基因组文库(由基因组DNA形成),以及随机突变体库(由被替代的核苷酸或密码子结合形成的新的基因合成)。基因文库是当前分子生物学的一个主要研究方向 ,这些文库的用途取决于原始DNA片段的来源。用于文库制备的克隆载体和技术存在差异,但通常每个DNA片段都被独立地插入一个克隆载体,然后将重组DNA分子转化入受体菌群(细菌人工染色体,BAC文库)或酵母内,使得每个生物体平均含有一个构建的表达载体(载体+插入物)。随着培养的种群的数量增长,其中包含的DNA也在被不断复制。
cDNA
Complementary DNA (cDNA) is a DNA copy of a messenger RNA (mRNA) molecule produced by reverse transcriptase, a DNA polymerase that can use either DNA or RNA as a template.
cDNA文库的制备
NGS数据格式介绍
一般情况下,从Illumina平台上得到的测序,其数据格式是Fastq格式,可以称之为原始数据(Raw data)。事实上直接的下机数据是显微拍摄得到的图像信息。但是一般都会用Bcl2Fastq软件将图像信息转化成Fastq文件。
如果测序是SE:则只有一个fastq文件,如果是PE测序,则得到两个Fastq文件。
PE的数据特点有:
(1)两个Fastq文件中分别包含数据1和2,来区分前后端;
(2)这2个文件的行数必须一致;
(3)相同的行上的数据来自同一条DNA片段双末端的测序数据;
(4)Fastq以每4行为一个单位,表示一条reads的信息。
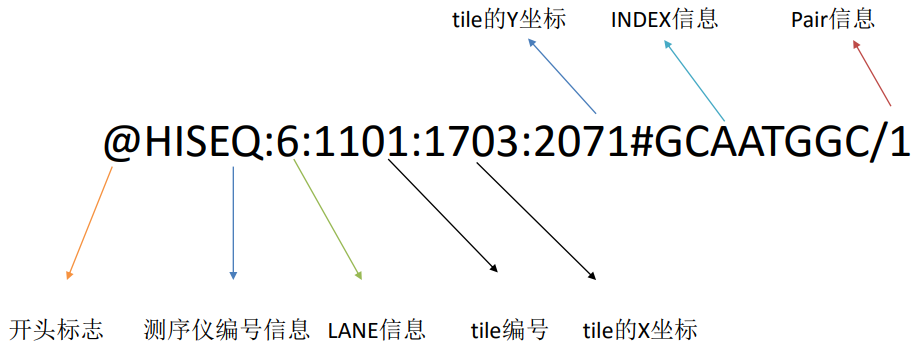
@HISEQ:6:1101:1703:2071#GCAATGGC/1
AGAATGCGTCATTCTGCGGAACTCATCCGACTGAATACCGAAAAGCAGAATCTGATCCTGGTTTCT
GCCATAAAGTAGCGCGAGCACACAGACGTCTGCGCGCCTGCGGTGACGGCGGTGCGGGT
````fdbeaeddf]d_ffNddPP\dedd]N[XPdffP\NeNdbff]faeafPdeff]PbPPP[efP^YePY\edfefO[
NNNbcM_effc\OcfcOWbffffMXcaMcffa_cYcYYbccYM]b
第一行 序列名称
第二行 序列的碱基组成
第三行 序列信息,或者直接以“+”做标记
第四行 碱基的质量
现在的Illimina使用的质量格式为Phred+33,和Sanger的碱基质量基本一致;碱基质量使用Q(Phred值)表示,其计算公式为:
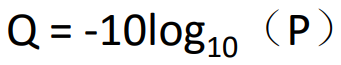
碱基质量与错误率的关系为:

CNS期刊
C:cell
N:nature
S:science
GWAS
全基因组关联研究(Genome-Wide Association Studies,GWAS)是指在全基因组层面上,开展多中心、大样本、反复验证的基因与疾病的关联研究,是通过对大规模的群体DNA样本进行全基因组高密度遗传标记(如SNP或CNV等)分型,从而寻找与复杂疾病相关的遗传因素的研究方法,全面揭示疾病发生、发展与治疗相关的遗传基因。
CPM
– Counts per million.
原始的表达量除以该样本表达量的总和,在乘以一百万就得到了CPM值 。从公式可以看出, CPM其实就是相对丰度,只不过考虑到测序的reads总量很多,所以总的reads数目以百万为单位。
假定原始的表达量矩阵为count, 计算CPM的代码如下
1 | cpm <- apply(count ,2, function(x) { x/sum(x)*1000000 }) |
UMI
– Unique molecular identifier. A randomized nucleotide sequence incorporated into the complementary DNA in the initial steps of RNA-seq protocol. Because this sequence is then carried in the subsequent amplification steps, it can be used to recognize multiple sequencing reads originating from the same physical mRNA transcript.
The usage of UMIs is recommended primarily for three scenarios: very low input samples, very deep sequencing of RNA-seq libraries (> 80 million reads per sample), and the detection of ultra-low frequency mutations in DNA sequencing. For many other types of projects, UMIs will yield minor increases in the accuracy of the data. In addition, UMI analysis is an excellent QC tool of library complexity.
- UMI序列,即unique molecular index,它是一段随机序列,也就是说每个DNA分子都有自己的UMI序列,UMI长为10bp,那么就有
4^10=1,048,576也就是100多万种变化。它的作用就是经过了PCR+深度测序后,找到reads与原始cDNA的对应关系 =》 每个DNA标签分子的ID号
它考虑到了这样一种情况:一个基因片段经过PCR扩增产生多个reads,但是不加标记我们是不知道的,并且不同基因的PCR扩增效率可能不同,因此一个基因最后得到的reads数就可能由于PCR扩增效率高而超过了另一个基因(而这两个基因的真实表达量可能差不多)。也就是排除”PCR bias”
Dropout
– In the context of scRNA-seq, this refers to a stochastic failure to detect a transcript that is expressed in a cell.
Sample Preparation Protocols
Broadly speaking, a typical scRNA-seq protocol consists of the following steps (illustrated in the figure below):
- Tissue dissection and cell dissociating to obtain a suspension of cells.
- Optionally cells may be selected (e.g. based on membrane markers, fluorescent transgenes or staining dyes).
- Capture single cells into individual reaction containers (e.g. wells or oil droplets).
- Extracting the RNA from each cell.
- Reverse-transcribing the RNA to more stable cDNA.
- Amplifying the cDNA (either by in vitro transcription or by PCR).
- Preparing the sequencing library with adequate molecular adapters.
- Sequencing, usually with paired-end Illumina protocols.
- Processing the raw data to obtain a count matrix of genes-by-cells
- Carrying several downstream analysis (the focus of this course).
PCR
聚合酶链式反应(英语:Polymerase chain reaction,缩写:PCR,又称多聚酶链式反应),是一项利用DNA双链复制的原理,在生物体外复制特定DNA片段的核酸合成技术。透过这项技术,可在短时间内大量扩增目的基因(标的基因)[1][2],而不必依赖大肠杆菌或酵母菌等生物体。DNA 体外扩增的实现首先基于DNA半保留复制原理,还有碱基互补配对原则,即复制的过程中双链DNA会解链,由双链变成单链,温度一般为95℃;之后温度降下来,引物会结合到DNA单链上,在DNA聚合酶的作用下,把游离的dNTP按照碱基互补配对原则结合到单链上,形成一条由旧链和新链杂合成的新的双链DNA。这个过程可概括为‘变性-退火-延伸’三个基本步骤。
内含子与外显子
内含子(英语:Intron)是一个基因中非编码DNA片段,它分开相邻的外显子。更精确的定义是:内含子是阻断基因线性表达的序列。DNA上的内含子会被转录到前mRNA中,但RNA上的内含子会在RNA离开细胞核进行转译前被剪除。在成熟mRNA被保留下来的基因部分被称为外显子。真核生物的基因含有外显子和内含子,是前者区别原核生物的特征之一。
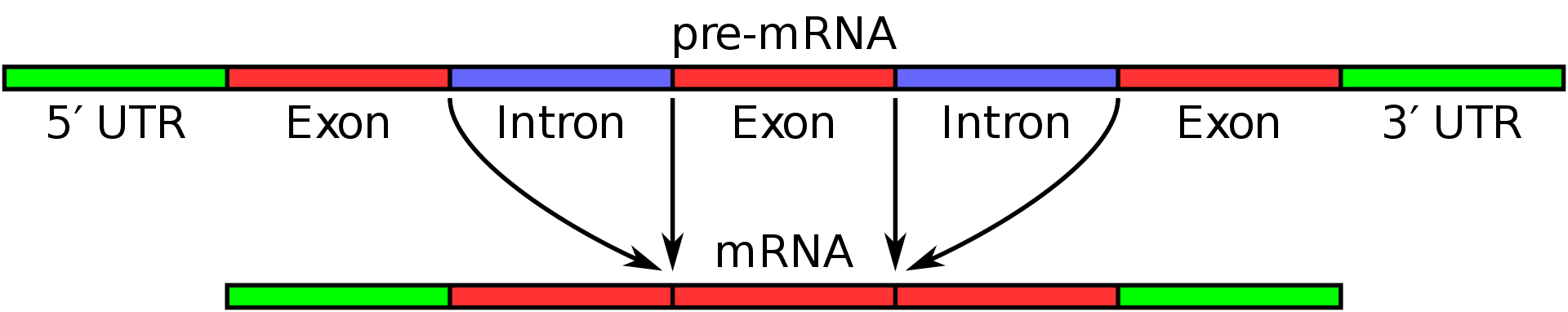
外显子:断裂基因中的编码序列。外显子是真核生物基因的一部分。它在剪接(splicing)后会被保存下来,并可以在蛋白质生物合成过程中被表达为蛋白质。外显子是最后出现在成熟RNA中的基因序列,又称表达序列。
外显子既存在于最初的转录产物中,也存在于成熟的RNA分子中的核苷酸序列。

剪接方式并不是唯一的(参看替代剪接),所以外显子只能在成体mRNA中被看出。即使是使用生物信息学方法,要精确预测外显子的位置也是非常困难的,外显子的识别及其拼接都是难题。 真核生物的基因,其线性表达被内含子阻断,这就是所谓的断裂基因(英语splitgene),该现象的发现者RichardJ.Roberts和PhillipA.Sharp获得了1993年诺贝尔奖。
RNA的种类
mRNA
依据DNA序列转录而成的蛋白质合成模板
tRNA
mRNA上遗传密码的识别者和氨基酸的转运者
rRNA
组成核糖体(蛋白质合成的机械)的部分
stem cell
干细胞(英语:stem cell)是原始且未特化的细胞,它是未充分分化、具有再生各种组织器官的潜在功能的一类细胞。干细胞存在所有多细胞组织里,能经由有丝分裂与分化来分裂成多种的特化细胞,而且可以利用自我更新来提供更多干细胞。对哺乳动物来说,干细胞分为两大类:胚胎干细胞与成体干细胞,胚胎干细胞取自囊胚里的内细胞团;而成体干细胞则来自各式各样的组织。在成体组织里,间叶干细胞与前体细胞担任身体的修复系统,补充成体组织。在胚胎发展阶段,干细胞不仅能分化为所有的特化细胞- 外胚层,内胚层和中胚层(参考人工多能干细胞),而且能维持新生组织的正常转移,例如血液、皮肤或肠组织。
四分位距(interquartile range,IQR)
四分位距,又称四分差。是描述统计学中的一种方法,以确定第三四分位数和第一四分位数的区别。与方差、标准差一样,表示统计资料中各变量分散情形,但是四分差更多为一种稳健统计(robust statistics)
Def:
四分位距通常是用来构建箱型图,以及对概率分布的简要图表概述。对一个对称性分布数据(其中位数必然位于第三四分位数与第一四分位数的算数平均数),二分之一的四分差等于绝对中位差(MAD)。中位数是集中趋势的反映。
公式:$IQR = Q3 - Q1$
举个例子:图表中数据
| 数列 | 参数 | 四分差 |
|---|---|---|
| 1 | 102 | |
| 2 | 104 | |
| 3 | 105 | Q1 |
| 4 | 107 | |
| 5 | 108 | |
| 6 | 109 | Q2 (中位数) |
| 7 | 110 | |
| 8 | 112 | |
| 9 | 115 | Q3 |
| 10 | 118 | |
| 11 | 118 |
从这个表格中,我们可以算出四分差的距离为 115− 105 = 10。
ERCC
External RNA Control Consortium (ERCC)
目前常用的ERCC版本内包含了176条RNA序列,这些RNA序列是从一些物种的DNA转录得到的,编号从ERCC-00001到ERCC-00176,所有对应的编号的序列全部可以从网上直接搜索下载。
目前,已经有非常成熟的商业化的ERCC序列,其中的176条RNA组成的一个库(pool)也是经过特殊设计的。不是简单的等量混合,而是不同的pool会对176条序列分成很多小库(subpool),然后通过不同的比例对这些subpool进行混合,然后形成不同的配比比例。在购买商业化的ERCC序列时,一定要注意下载对应版本的绝对定量信息。当然,有些商业化的ERCC里面也不是包含全部176条标准序列。
https://zhuanlan.zhihu.com/p/52063724
transcript
转录本是由一条基因通过转录形成的一种或多种可供编码蛋白质的成熟的mRNA。
也称为剪切体
一条基因通过内含子的不同剪接可构成不同的转录本。设计转录本实验可以研究内含子剪切机制、表观遗传、RNA编辑等,通常是考察一条基因对应的不同转录本的调节机制等。
QC的指标
三种QC指标(the number of genes、the count depth 、the fraction of mitochondrial reads)要放在一起思考,而不是单独看某一个
测多少数据量?几个G?多少reads?如何换算?
根据研究目的决定测序深度:
目的1:通过抓取polyA尾巴建库(只测那些带有polyA尾巴的基因,大多是蛋白编码基因),
寻找样品间基因转录谱的相似性,只需要30M reads,长度大于30nt即可,双端测序,其中20-25M能够回帖到已知转录组上。
目的2:要发现新的转录本,对已知isoform(同一基因由于不同的可变剪接方式形成多种isoform,勉强译为亚型)进行定量分析,
兼顾低表达量的转录本或isoform,就需要100-200M read,长度大于76bp,双端测序。
lncRNA-seq属于这一类型。
注:ENCODE测的是人和小鼠,其他物种不包括在此推荐范围内。
另外,miRNA测序,只需要10M read,每条read长50bp,单端测序。
ChIP-seq,需要20M read,每条read长50bp,单端测序。
销售只说多少G,不说reads数,如何把reads数换算成G呢?
这跟测序长度有关:
PE150或2*150,即 双端测序,每条read长度150bp。
150bp X 2端 X read数 = 数据量
例如,测50M read,150bp X 2端 X 50M read = 15000M = 15G
注:对于双端测序,一个RNA片段,即fragment,也叫read,会测出来2条序列。
SE50或1*50,即 单端测序,每条read长度50bp。
50bp X 1端 X read数 = 数据量
例如,测20M read,50bp X 1端 X 20M read = 1000M = 1G
再絮叨一句:这里的G是碱基数(Gbase,Gb),跟你看到的文件大小(gigabyte,GB)不是一回事哦~
测序公司给你的文件通常是压缩的fastq格式,里面有read ID号,有碱基,有每个碱基的质量。
小哈看到文件大小就感觉数据量不够,是基于经验的推测,要明确测了多少数据量,跑一个FastQC或RSeQC就知道了。
差异基因表达时log2FC和FDR值含义
log2FC中的FC指fold change,表示两组样品间表达量的壁纸,对其取2为底的对数之后即为log2FC。一般,默认取log2FC绝对值大于1为差异基因的筛选标准;
FDR即False Discovery Rate,错误发现率,是通过对差异显著性p值(p-value)进行校正得到的。由于转录组测序的差异表达分析是对大量的基因表达值进行独立的统计假设检验,会存在假阳性问题,因此在进行差异表达分析过程中,采用了公认的Benjamini-Hochberg校正方法对原有假设检验得到的显著性p值(p-value)进行校正,并最终采用FDR作为差异表达基因筛选的关键指标。一般取FDR<0.01或者0.05作为默认标准。